肺高血圧症

千葉大学大学院医学研究院
呼吸器内科学
千葉大学病院
呼吸器内科
特発性肺動脈性肺高血圧症 (idiopathic pulmonary arterial hypertension: IPAH)
遺伝性肺動脈性肺高血圧症 (hereditary pulmonary arterial hypertension (HPAH) )
(医療従事者向け) 2014年7月
現在の肺動脈性肺高血圧症の分類は「肺高血圧症の歴史」述べているようにニース分類に則っている. 遺伝子素因が明らかなものは”Heritable PAH”, 膠原病, HIV感染, 門脈圧亢進症や先天性心疾患など他疾患に続発して発生する肺動脈性肺高血圧症は”Associated with PAH (APAH)”に分類される. こうした明確な原因疾患が除外され, “原因不明”として残った疾患群が”特発性肺動脈性肺高血圧症(IPAH)”と診断される(図1).
注:上記のような疾患概念及び定義の変遷を経ており, ”PPH”は”IPAH”と未分類であった”HPAH”が混在し, 従来の”PPH”についての報告を”IPAH”と読み替えることは出来ない. したがって本稿では両者の記載が混在することをご了承下さい.

肺性心の剖検例のうち約1%にPPHが認められたとの報告があるが, PPH (IPAH/HPAH)の正確な発生頻度は不明である. 本症は右心カテーテル検査による確定診断が必要であるが必ずしも全ての施設で行える検査とはいえず, 確定診断まで至らない症例も潜在的に存在すると考えられる.
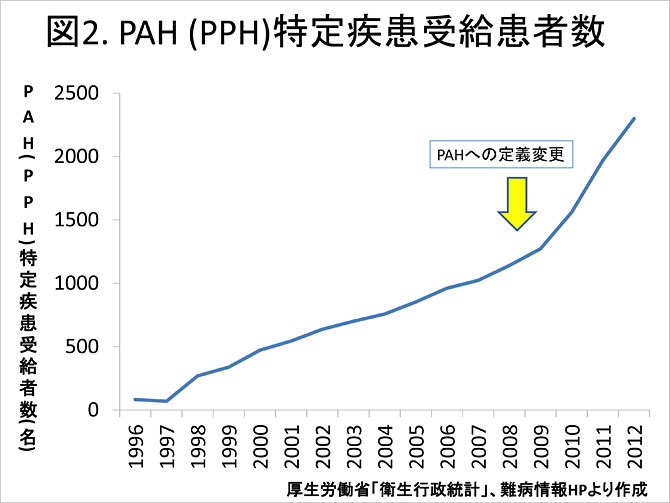
1997年に行われた本症の全国疫学調査では, 当時の改訂PPH診断基準での全国推計患者数は230人(95%信頼区間200~226人)とされた. PAH (PPH)特定疾患受給患者数は年々増加し, 2012年ではおよそ2,300人に達している(図2).
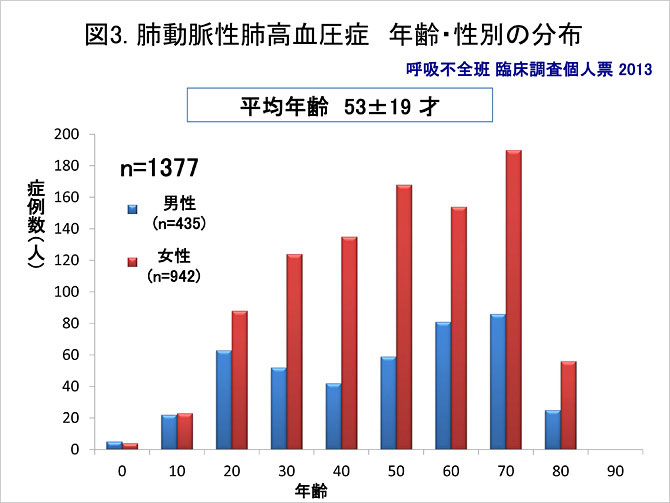
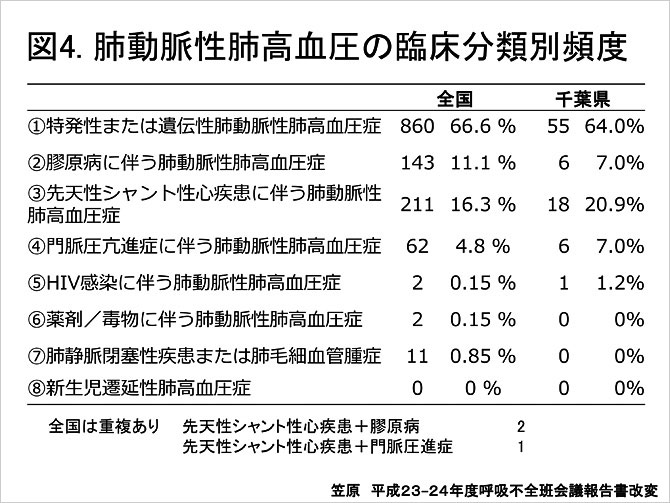
厚生省特定疾患呼吸不全調査研究班におけるPAHの臨床調査個人票の解析では, 成人PAHは女性に多くみられ, 男女比は約1:2.17とされている (図3). 発症年齢は比較的広く分布しているが, 50歳から70歳までにピークが見られる. 以前は20-40歳代と比較的若年層に多いとされてきたが, 有効な治療法の確立により治療予後が改善したことなどから(1), 以前の年齢ピークが高齢側にシフトしたことが示唆される. 分類別の統計では特発性または遺伝性肺動脈性肺高血圧症が最多となっている (図4). この統計は肺動脈性肺高血圧症特定疾患申請書類を基に集計されており, 膠原病に対する特定疾患受給を受けている患者は上記統計に含まれていない可能性から結果の解釈には注意が必要である.
本症の病変部位は, 毛細血管より上流側に位置する末梢肺小動脈である. 病態の中心は, こうした末梢肺動脈の収縮, リモデリング, 二次的な血栓形成などによる肺動脈の狭小化とそれに続発する肺血管抵抗の著明な増加といえる(Heart View 2011;15:33).
血管収縮物質/血管拡張物質の不均衡
PAH患者においては肺動脈内皮細胞において肺血管収縮物質であるエンドセリン-1が過剰発現する一方 (N Engl J Med 1993;328:1732), 血管拡張物質であるプロスタサイクリン合成酵素の発現低下 (Am J Respir Crit Care Med 1999;159:1925)が報告され, 血管収縮物質と血管拡張物質の不均衡による肺血管攣縮がPAHの病態発生の首座であると考えられていた (N Engl J Med1992;327:70). PAH病態発生に関しては後述する肺血管リモデリングの探索にシフトしてきているが, 肺血管攣縮に関わるとされるこれらの分子は現在肺血管拡張薬の主なターゲットとなっており, その重要性は薄れていない (2).
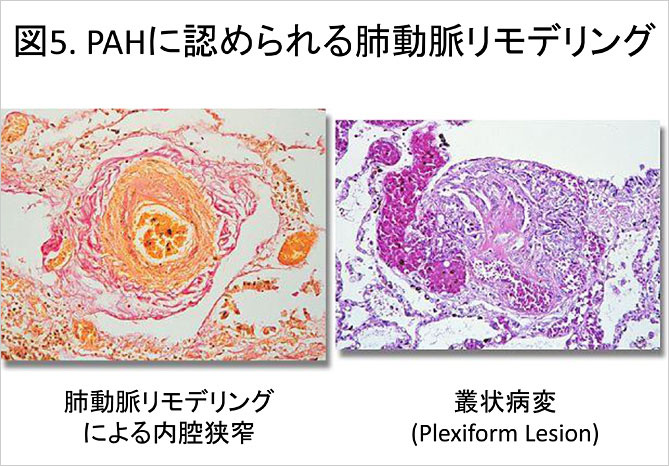
IPAH患者の肺組織には肺動脈のリモデリング所見が病理学的に認められることが古くから知られていた. 病理組織学的には肺動脈中膜肥厚, 内膜の同心円状線維化等の肺動脈リモデリングの他, 血管の内腔と既存構造の破壊を伴った叢状病変(Plexiform lesion)等が認められる (図5). 肺動脈リモデリング部位ではアポトーシス抵抗性内皮細胞が増殖していることが報告され(3), 近年血行動態とは無関係に増殖し一部で悪性細胞と似た細胞増殖性疾患であるとの疾患概念の大きな転換が行われている(Cancer paradigm) (Am J Respir Crit Care Med 2008;178:558).
なお, こうした肺血管リモデリング所見はIPAHに特異的な所見ではない.膠原病に伴う肺高血圧症(Human Pathology 2007;38:893)の他, 慢性血栓塞栓性肺高血圧症(Chest. 1993;103:685)など他グループに分類される肺高血圧症でも報告され, 肺高血圧症に属する疾患群では共通してみられる病理学的所見と言える.
IPAHの成因に関してはいまだ明らかではない. 従来PPHの約6%に家族内発症が認められることが知られており(Rich 1987)何らかの遺伝子的要素の関与が示唆されてきた. 近年いくつかの遺伝子異常の関与が報告され, 遺伝性肺動脈性肺高血圧症(HPAH)と分類される病態が増えてきている. PAH診断基準に記載されている遺伝子異常としてBone morphogenetic protein receptor type II(BMPR2)とActivin receptor-Like Kinase1(ALK-1)などが挙げられる.
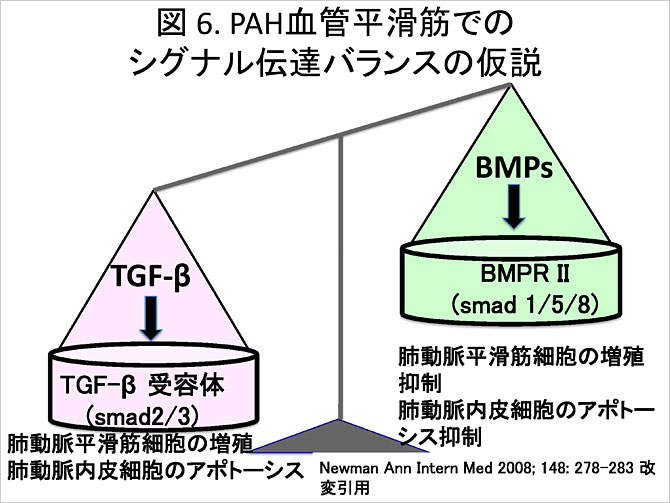
BMPR2遺伝子異常は2000年にPPH発症との関連を見出された(Am J Hum Genet 2000;67:737). BMPR2遺伝子異常によりtransforminggrowth factorβ (TGF-β)系のシグナル伝達が強くなる結果, 肺動脈における平滑筋細胞の増殖と肺動脈内皮細胞の過剰なアポトーシスが誘導される(Ann Intern Med 2008;148:278) (図6). BMPR2遺伝子異常は家族性PAHの50-100%, 孤発性のIPAHの約25%において陽性とされるが, 本遺伝子異常を持っていてもPAHを発症する症例は10-20%に過ぎない(Respiration 2007;74:123). これらの遺伝子要因に加え, 環境要因などの何らかのトリガーが加わって発症すると考えられている(Heart View 2011;15:26).
ALK-1遺伝子異常は, 元々遺伝性出血性毛細血管拡張症(HHT)における肺高血圧症への関与が指摘されていたが (N Engl J Med. 2001;345:325), HHTのないPAH患者にも同遺伝子変異が存在する事が判明し, PAH関連遺伝子として認められるようになってきた (J Am Coll Cardiol. 2009;54:S32). 発症機序としては TGF-β伝達系の障害との関連が示唆されてきている (N Engl J Med. 2001;345:325).
PPH患者80例の病理学的検討では20-56%の患者で微小肺血栓が認められ, PPH発症との関与が指摘されている(Mayo Clin Proc. 1985;60:16). 背景となる凝固異常として血小板凝集能の亢進, プラスミノーゲンアンチベーターインヒビター上昇, トロンボモデュリンの低下の報告がみられる(日本臨床2001;59:1053).
左室から拍出された血流が多くの臓器へ還流する体循環と異なり, 肺循環では右室から拍出された血液は全て肺に流入する. したがって, 肺動脈圧や肺血管抵抗の上昇により増加した後負荷の影響は右室がダイレクトに受けることになる. 右室に対する後負荷の持続的かつ進行性増大に対して, 右室筋は肥大することにより収縮力を強め右室拍出量を維持する. しかしながら, 後負荷の増大が右室の限界以上もしくは進行が急激な場合, 右室は適応不能に陥る. 代償不可能となった右室および右房は拡張をきたし右心不全状態が惹き起こされる(図8)(J Am Coll Cardiol 2013; 62: D22).
一般に, 右室に対する血流抵抗の増大により右室拍出量が制限されるため, 心拍出量は低く, 同時に体血圧も低値を示すことが多い. このため, 体動などにより四肢の筋肉への血流が増加すると, 脳血流の減少から失神をきたすこともある. 本症では, 動脈血酸素分圧(PaO2)は正常か軽度の低下にとどまることが多い. 本症での低酸素血症の要因としては, 低心拍出量による混合静脈血酸素分圧の低下に加え, 肺毛細血管レベルでの肺胞気との接触時間の短縮なども考えられている. しかしながら, 右房の拡大や右房圧の上昇により卵円孔の再開通が生じると, 心内右左シャントにより著明な低酸素血症をきたすことがあり注意が必要である.
自覚症状として肺高血圧症に特異的なものはない. 労作時呼吸困難はPAHの初発症状として出現することが最も多く, 診断確定時にはほぼ全例に認められる. このほか, 易疲労感や倦怠感, 胸痛, 失神 などを症状として訴えることが多く, 病気の進展に伴い右心不全の合併による症状が加わってくる (図8). また, 稀ではあるが, 左肺動脈の拡大により左反回神経麻痺が生じ, 嗄声が認められることもあり注意が必要である. 一般に, 初発症状出現から診断確定までの期間は平均2-3年とされている.

理学所見としては, 頻脈, 手足の冷感, チアノーゼなどに加え, 右心不全が合併すると, 下腿浮腫や肝腫大などの所見が認められる. 胸部聴診上では, II音の肺動脈成分の亢進が最も高率に認められ, このほか右心性のIII音, IV音も聴かれる場合がある. また三尖弁逆流が生じると収縮期心雑音が, 肺動脈弁逆流がおこると拡張期心雑音も聴かれるようになる. さらに, 肺高血圧症が著明になると, 第2肋間胸骨左縁にて肺動脈の拍動が触診さらには視診にて確認できるようになる.
PAHの診断は厚生労働省診断基準に依る. 臨床症状および検査所見などにPAHに特異的とされる所見は少ない. したがって他の原因による肺高血圧症の除外が診断のポイントといえる.
血液検査上PAHに特異的な血液検査所見はなく, 二次性の肺高血圧症の除外の側面が強い. 赤沈値亢進, γ-グロブリン増加, 抗核抗体陽性, 血清補体価低下や各種自己抗体の測定などが膠原病に伴う肺高血圧症の発見契機となる事がある. 肝機能検査異常を示した症例では肝硬変や門脈圧亢進などの肝疾患による肺高血圧症の存在を念頭に置く必要がある.
動脈血ガス分析では, PaO2の軽度低下と同時に, PaCO2の低下も認められる事が多い. しかし動脈血液ガスが正常の症例や,右心負荷の増大にともなう卵円孔再開通の結果生じた逆シャントのためPaO2が著しく低下する症例も存在し, 症例によるばらつきが認められる.
心拡大などに伴う軽度の肺活量の低下のほか, 肺拡散能の中等度以上の低下が認められる場合が多い.
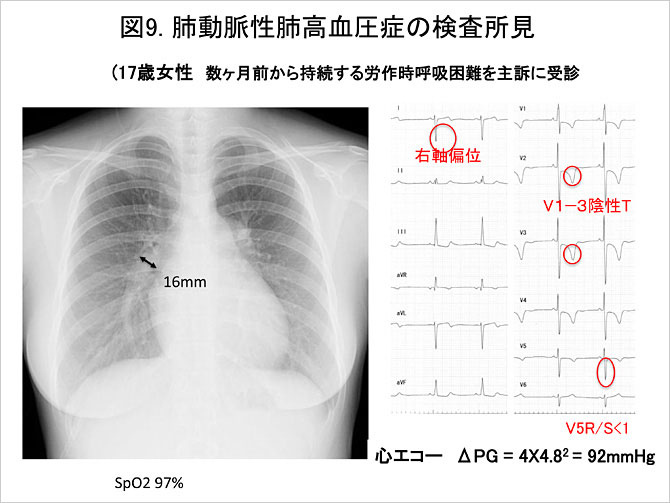
肺高血圧症に伴う右室肥大, 右房負荷所見を捉える. 100°以上の右軸偏位, V1でのR/S>1, V5でのR/S<1といった右室肥大の所見に加え, 肺性P波などの右房負荷所見などが認められる(図9).
左第二弓の突出や右肺動脈下行枝の拡大に加え, 心拡大などが認められる(図9). 一般にPAHでは末梢肺血管陰影が減少し, 肺野末梢が明るく見えることが特徴とされ, これが心内シャント性疾患との鑑別点の1つともなっている.
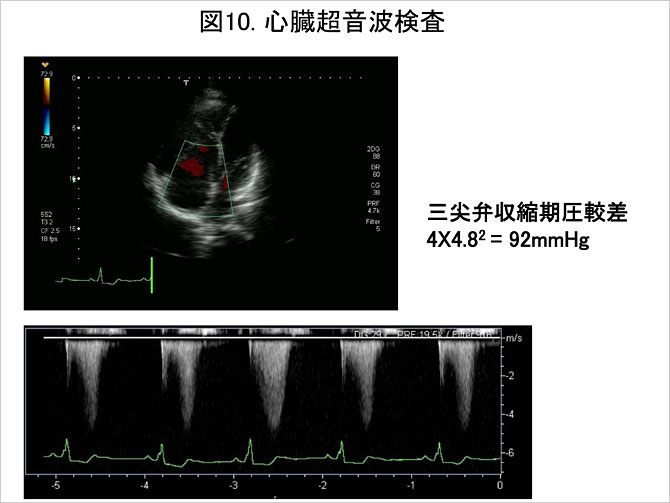
心臓超音波検査は肺循環動態の評価やフォローアップにおける肺動脈圧の指標を提供する重要な検査である. 右室・右房の肥大および拡大に加え, 心室中隔の左室側への圧排や奇異性運動などが観察される. また,ドップラー法応用により, 三尖弁および肺動脈弁での逆流波の流速測定から, 弁前後での圧較差の測定および収縮期肺動脈圧の推定が可能である. また心エコーは先天性心疾患に伴う肺高血圧症および各種心疾患に伴う後毛細血管性肺高血圧症を否定する意味でも不可欠の検査といえる.
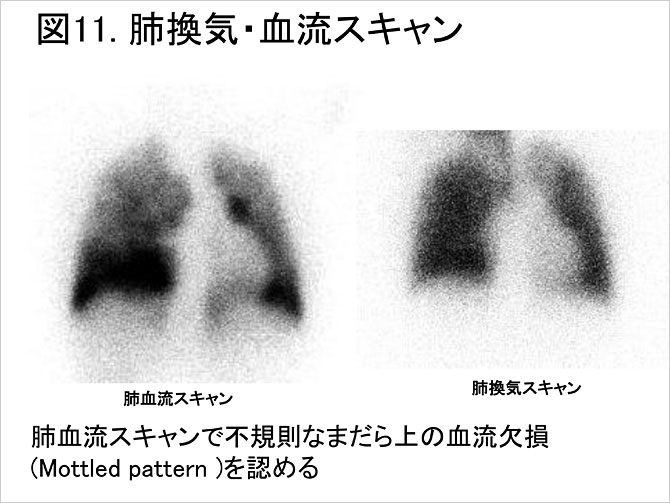
PAHでは, 肺血流スキャンは正常もしくは解剖学的区域に一致しない不規則な斑状の血流欠損像を示すとされる(図11). 後述する慢性血栓塞栓性肺高血圧症では血流欠損のパターンが区域レベルの血流欠損像を呈することが鑑別点ともいえる. また, 肺換気スキャンは正常である.
肺動脈主幹部の拡大, 心室中隔の扁平化ないしは左室側への圧排などが認められる. また, 造影CTにて肺動脈中枢側に血栓像のないことを確認しておくことも重要である.
右心カテーテル検査は肺高血圧の程度ならびに心拍出量などの肺循環動態の正確な評価を行う必須の検査である. 前毛細血管性肺高血圧症の確認のため, 肺動脈平均圧が25mmHg以上であり, かつ肺毛細血管楔入圧が12mmHg以下の正常値を示すことを確認する. さらに, 先天性心疾患による心内シャントがないことを確認することも診断上重要である. IPAH では重症例を除いては, NOを用いた肺血管拡張試験を行う.
詳しい除外診断および組織学的診断確定のため, 肺組織の病理学的診断を加える場合もある. しかし侵襲が大きいこと, 病理学的所見が必ずしも治療方針と結びつかないこと, 肺移植が必要となった場合に胸膜癒着が不都合となることなどから最近では推奨されない (肺高血圧症ガイドライン 2012年)
CTEPHはPAHと異なり外科的治療(肺動脈血栓内膜摘除術:PEA)が可能であることから, その鑑別は臨床的に極めて重要である. PAHとの鑑別のポイントは肺動脈内血栓存在の有無を示すことにある. 肺高血圧症が存在する症例で肺血流スキャンでの区域枝以上の血流欠損所見や造影CTによって肺動脈内血栓が認められる場合は, 慢性血栓塞栓性肺高血圧症を強く疑う. 一方, 正常あるいは 非区域性斑状欠損の場合PAHが示唆される. さらに, 肺血流スキャンが正常の場合, 中枢肺動脈に血栓あってもCTEPHではなく, PAHに血栓が合併した可能性が高い. PAH患者に対する肺動脈造影検査は検査による危険性もしばしば報告されており, その実施にあたっては適応を含め十分な注意が必要である.
皮膚症状や関節症状の有無, 赤沈亢進, CRP高値などの炎症所見の有無を参考に, 各種自己抗体の検査などからそれぞれの膠原病診断基準に当てはまるかどうか検討する. PAHにおいても, 抗核抗体陽性やγ-グロブリン値増加など膠原病に類似した血液検査所見が認められ, いずれの膠原病診断基準をも満たさない場合にはIPAHと臨床的に診断される.
血液検査における肝機能異常の有無や腹部超音波エコーにて肝硬変の有無を確認する. また, ドップラーエコーによる門脈血流方向の確認や, 門脈-下大静脈シャントの有無などを参考に門脈圧亢進の有無を診断する. 症例によっては, 確定診断のため門脈造影などが必要なこともある.
高安病に伴う肺動脈炎と特発性のものが知られているが, 高安病に伴うものでは, 脈の左右差や特徴的な眼底所見が鑑別点となる. 肺血流スキャンでは, 肺区域枝以上のレベルでの血流の低下ないしは欠損像がみられ, 肺動脈造影では中枢側の比較的太い肺動脈に狭窄像や閉塞像が認められることが多い.
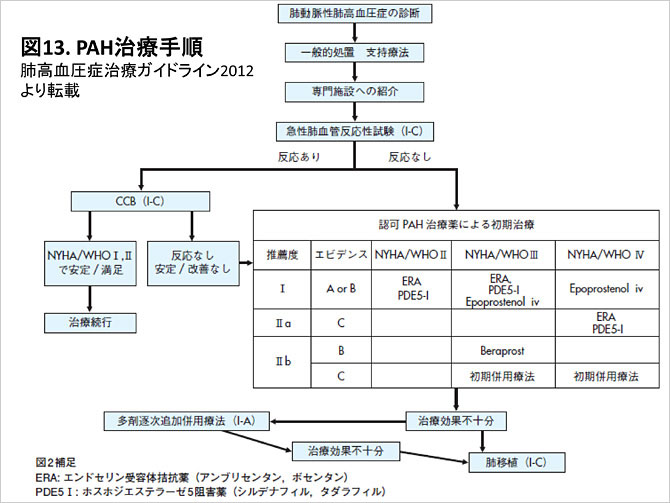
本症の治療は抗凝固療法, 酸素投与, 右心不全対策の生活指導などの支持療法を前提とした上で, 急性肺血管反応試験の結果や重症度に応じて肺血管拡張薬の適応を決定する (図13).
A. 支持療法
本症の進行過程には, 微小血栓が関与していると考えられており(Mayo Clin Proc. 1985;60:16), 過去の後ろ向き試験で生命予後に対する有用性が報告されている(Circulation 1984;70:580, N Engl J Med 1992;327:76). したがって, 原則として全例で抗凝固療法を施行する.
PPHでは 動脈血酸素分圧および混合静脈血における酸素飽和度(PaO2およびSvO2)と生命予後が極めて強く関連することが報告されている(Circulation 1984;70:580) SvO2の低下は末梢組織レベルの組織低酸素を反映し, 心拍出量の低下と関連していると考えられる. 本症では安静時低酸素血症が認められない症例でも積極的に在宅酸素療法を施行する.
過労・ストレスを避け, 水分・塩分のとりすぎを避けるなど右心不全を予防する. 右心不全症状出現時は, フロセミド等の利尿薬を基本とし, 適否に問題はあるものの頻脈等のみられる場合強心薬も使用される. 右心不全が増悪し低心拍出量・低血圧のみられる場合, ドブタミン (3~10μg/kg/min)やドーパミン(3~10μg/kg/min)を使用する.
B. 肺血管拡張療法
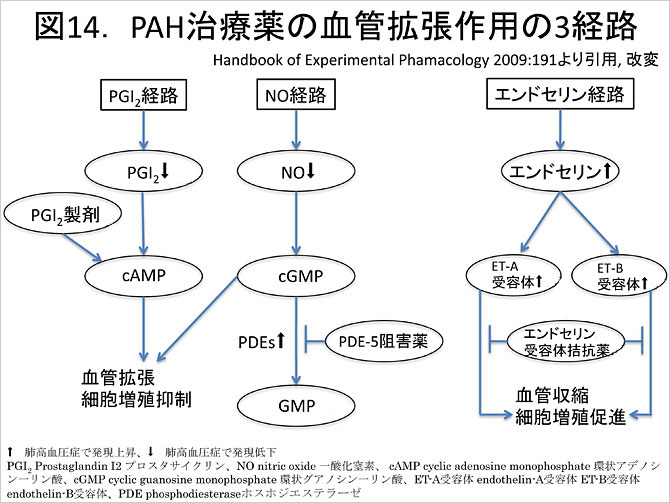
カルシウム拮抗薬は細胞膜に存在するカルシウムチャネルに結合し細胞内へのCa2+流入を阻害することで, 平滑筋の弛緩を促すという点で血管拡張の機序としては体血管の拡張機序と共通している. 肺血管に特有の分子をターゲットにした肺血管拡張薬の作用経路は大きく三つ想定されている (図14)
肺血管拡張薬についてのメタアナリシスではPGI2, エンドセリン受容体拮抗薬とホスホジエステラーゼ-5阻害薬のPAHに対する有効性が証明されている(Respir Res 2010;11:12). 最近では重症例を中心にこれらを組み合わせた同時併用療法の有効性が報告されている(4).
欧米ではカルシウム拮抗薬による大量療法の有用性が報告され (N Engl J Med 1992;327:76), 肺血管反応試験陽性例で適応となる. 急性肺血管反応性試験には主に一酸化窒素(nitric oxide: NO)が用いられる(図15). 10-20ppmのNOを10分間吸入し,「吸入前と比較し10mmHgの平均肺動脈圧低下」かつ「吸入後平均肺動脈圧10mmHg以下」が陽性の条件となる(Eur Heart J 2009;30:2493). 正確な頻度は不明であるが本邦ではこの陽性率が低く,また大量療法に耐えられる症例は多くないのが現状である.
PGI2は1976年に発見された強力な血管拡張作用のある内因性生理活性物質である (Nature. 1976;263:663). 作用機序として, PGI2製剤は血管平滑筋のPGI2受容体に結合し, 細胞内のcAMP濃度を増加させ平滑筋を弛緩させる. この他, 血小板凝集, 粘着能抑制作用による肺動脈内血栓形成抑制, 血管における細胞増殖やリモデリング抑制作用の可能性も報告されているが, これらの詳細は不明である.
きわめて予後の悪い疾患であったPPHに対しPGI2製剤であるエポプロステノールが1980年代から臨床的に用いられ, 1980年代から1990年代にかけ臨床試験が行われその有効性が示されてきた. 最近のメタアナリシスではPGI2製剤による生存への寄与, 運動耐容能の改善などPAHに対する様々な有効性が証明されている(Respir Res 2010;11:12). 急性効果で血管拡張反応が不良であった症例においても, 長期効果での有用性が得られている.
PGI2製剤に共通した有害事象としては頭痛, 熱感, 紅潮, 消化器症状などがある.
ベラプロストは本邦では肺高血圧症患者に対し広く使用されている薬剤である. 本邦の2012年版肺高血圧症ガイドラインではWHO機能分類III症例に対してのエビデンスレベルはClass II-bとなっている.
ベラプロストは本邦から平均肺動脈圧や総肺血管抵抗(TPR)の改善が報告され (Lancet 1997;349:1365), 1998年PPHに保険適応となった. その後長時間の血中濃度の維持を目的とされベラプロスト徐放製剤が開発された. PAH患者に対する臨床第2相試験ではベラプロスト徐放製剤が平均肺動脈圧, 総肺血管抵抗, 6分間歩行距離の改善を示し, 血行動態の改善及び運動耐用能の改善が報告されている (Int Heart J 2009;50:513). アメリカの報告では長期効果が明確ではなかったこともあり(J Am Coll Cardiol 2003;41:2119), 長期効果に対してはさらなる検討が必要である.
エポプロステノールのPAHに対する有効性はメタアナリシスを含め肺血行動態改善および生命予後改善効果が報告され(Ann Intern Med 1994;121:409, Circulation. 2002;106:1477, Respir Res 2010;11:12), 肺高血圧症治療薬として高いエビデンスを持つ治療薬である. 本邦の肺高血圧治療ガイドライン2012年版でもWHO機能分類 III 度, IV度例ではエポプロステノールの適応とされる (エビデンスレベルIA or IB). PGI2治療によるPAHの予後が明らかに改善したため, PGI2治療前の肺血行動態によって患者の予後を予測することは困難となっている. 治療の指標としてはヒト脳性ナトリウム利尿ペプチド(BNP)等を用いる.
エポプロステノール製剤は水溶液で強アルカリを示し, かつ健常人での血液中の半減期は6.3分程度(Thromb Res. 1986;43:379-87)と極めて短いことから, 中心静脈カテーテルから持続投与することが必要となる. その際他の薬液と混ざらないよう配慮が必要となる. 現在は, 0.5~1ng/kg/minより開始し, 以後自覚症状と臨床症状の改善を目安にゆっくり増量する(0.5~2 ng/kg/minを3日~1週間毎に)のが安全と考えられているが, 増量の程度や最大投与量などについての専門医の見解は確定していない. エポプロステノールは本来常温環境下において8時間ほどで失活してしまうことから, アイスパックなどによる薬剤の常時冷却も必要であった. しかし常温でも安定なエポプロステノール製剤が開発され, 2013年6月より販売され使用されている.
外来治療においては, 長期血管内留置用のカテーテル(Hickman皮下トンネルカテーテル)と携帯用小型ポンプを使用する. 副作用としての低血圧に対する対処, さらに留置カテーテルによる感染症などの合併症にも注意が必要であり, 患者教育が極めて重要である.
皮下投与プロスタグランジン製剤であるトレプロスチニルの有効性が報告され(Am J Respir Crit Care Med 2002;165:800), エポプロステノールと同様の効果が報告されている (J Heart Lung Transplant 2013;32:889). 本邦でも臨床第2相, 第3相試験での有効性, 安全性が示された (Progress in Medicine 2014; 34:333). 本邦では2014年7月現在保険収載されていないが今後本邦でのPAH治療の選択肢となる可能性がある. 本邦でも2014年承認され, 近日薬価収載予定である. 今後本邦でのPAH治療の選択肢となる可能性があるだろう.
エンドセリン受容体拮抗薬はメタアナリシスで血行動態の改善に加え6分間歩行距離およびBorg scaleの改善, WHO機能分類の改善が示されており(Respir Res 2010;11:12), WHO機能分類 II, III度の症例で選択される (図14. 肺高血圧症治療ガイドライン 2012年版). エンドセリン-1 (ET-1)は血管平滑筋に発現するエンドセリンA及びB受容体に結合し, 血管収縮や細胞増殖に促進的に働く(Pharmacol Res 2011;62:463). 肺高血圧症患者においては肺動脈内皮細胞において過剰発現していることが知られ (N Engl J Med 1993;328:1732), 肺高血圧症の病態形成に重要な役割を果たしていると考えられている. エンドセリン受容体拮抗薬はエンドセリン受容体に対する阻害作用により肺血管拡張をえるというものである. 本邦で現在保険収載されているエンドセリン受容体拮抗薬はボセンタンとアンブリセンタンである. ボセンタンはエンドセリンA受容体とB受容体両方の拮抗薬であり. アンブリセンタンはエンドセリンA受容体の拮抗薬である.
新しいエンドセリン受容体拮抗薬マシテンタンが注目されている. PAH患者を対象としたプラセボ対照無作為二重盲検比較試験 (SERAPHIN試験)でマシテンタンが有意に生存を改善させることが示されている (N Engl J Med 2013;369:809). さらに肝臓への蓄積性がないことから(Eur J Pharmacol. 2013;701:168), エンドセリン受容体拮抗薬の有害事象として懸念される肝障害も同試験ではプラセボと差が認められなかった. 以上の結果より2013年11月アメリカFDAで承認され, 本邦でも臨床試験が進行中である.
現在本邦で保険収載されているPDE-5阻害薬はシルデナフィルとタダラフィルである. 2010年のメタアナリシスでは肺動脈圧及び肺血管抵抗の低下など血行動態の改善に加え6分間歩行距離の改善など運動耐容能の改善も示されている (Respir Res 2010;11:12).
肺血管平滑筋に多く存在するPDE-5はcyclic GMPの分解酵素であるが, PDE-5阻害薬はこれを阻害することで細胞内でのcyclic GMPの濃度を高く維持する (図15). cyclic GMPは細胞内のCa2+濃度を低下させることで肺血管を拡張させる (Heart View 2011;15:60). 網膜色素変性症の3-4%の患者にPDE6Aの遺伝子異常が認められること(Retnet: https://sph.uth.edu/retnet/) が知られており, PDE5阻害薬による網膜色素変性症に対する影響が否定できない. そのため本邦での臨床試験では除外患者とされ, こうした患者に対するPDE5阻害薬の使用は「慎重投与」となっている. PDE5阻害薬の使用開始に当たっては眼科コンサルトの上で上記疾患の除外が必要と考えられる.
PAH患者に対する臨床第2相試験では12週のリオシグアト投与後肺動脈圧及び肺血管抵抗など血行動態が改善したことに加え, 6分間歩行距離がベースラインである370mから57mの優位な改善を認めた (Eur Respir J 2010;36:792).可溶性グアニル酸シクラーゼ(sGC)を直接刺激しcyclic GMPの合成を促す薬剤がリオシグアトである. PDE-5阻害薬とは細胞内cyclic GMPを増加させる機序が共通しており, リオシグアトとの併用は禁忌とされる.
現在本邦では手術不能末梢型CTEPH及びPEA術後に肺高血圧が残存したCTEPH患者に対して保険適応とされている. 海外ではすでに承認されており, 日本でも承認申請であり, PAHに対する治療選択肢となり得る.
Rhoキナーゼは細胞内Ca2+濃度非依存性に血管平滑筋の収縮や弛緩を制御することが知られ, Rhoキナーゼ阻害薬の肺高血圧症としての可能性が注目されている. 当科ではBMPR2異常により発症する肺高血圧症モデルマウスにおいてRhoキナーゼが肺動脈圧の上昇, 肺動脈リモデリング, 肺毛細血管床の減少等を抑制することを報告した(5). また, PAH患者における肺動脈ではRhoキナーゼの発現及び活性が亢進しており, 肺動脈の過収縮がRhoキナーゼ阻害薬に依って抑制されることが報告されている(Circ J. 2009;73:1731).
PAHは本邦で施行されている脳死両肺移植の適症疾患で最多である(移植 2013;48:374). 肺高血圧症における肺移植の適応基準は以下の通りである (図16). PGI2持続静注療法を始めとする最大限の内科治療にもかかわらず, 病態の改善がみられない症例が肺移植の適応となる. 肺移植認定施設での適応検討, 中央肺移植適応検討委員会での承認を受けた後, 日本臓器移植ネットワークに登録する手順となっている. なお術式については本邦ではその安全性からPAHについては, 両側片肺移植が標準術式となっている. ドナー不足の現状から, 近年, 岡山大学を中心に, 生体部分肺移植が試みられ, 良好な成績が報告されている.

今から約25年前は無治療例ならば診断確定からの中間生存期間は2.8年, 5年生存率34%程度とされ極めて予後不良の病態であった(Ann Intern Med. 1991;115:343). しかし上述のような診断法の確立や治療法の進歩によって予後の大幅な改善がみられるようになっており, 適切な診断・治療が極めて重要である. 死因としては, 右心不全が約50%と最も多く, このほか突然死も約25%にみられ注意が必要である.
日常生活での注意点としては, 肺動脈圧を上昇させる可能性のあるものは避けること, および右心不全症状を悪化させないことが肝要といえる. 過度の運動は, 心拍出量の増大から肺高血圧を悪化させ右心不全や不整脈を招くことがあるため控えさせる. 喫煙および高所への旅行も, 肺動脈圧の上昇をきたすため避けるように指導する. また, 妊娠および出産を契機とした病態の悪化がしばしば報告されており, 一般に妊娠は避けることが望ましい. PGI2療法使用例では, 急速注入や停止によって, 低血圧やショックをおこすこともあることから, 近医や訪問看護, 在宅管理システムとの連携をはかることが重要である. また, ポンプ不具合や留置カテーテルによる感染症などの合併症に対処する. このほか, 水分の摂取量ならびに尿量を毎日チェックする習慣をつけさせ, 体重も毎日ほぼ同じ時間に測定するように指導する. 体重の増加や尿量の減少に加え, 下腿の浮腫などが認められた場合, 安静度を強めるとともに利尿剤の追加内服を行わせ, 右心不全状態からの早期離脱を目指す.